Medical Regulatory
Clinical Evaluation/Trials Approval
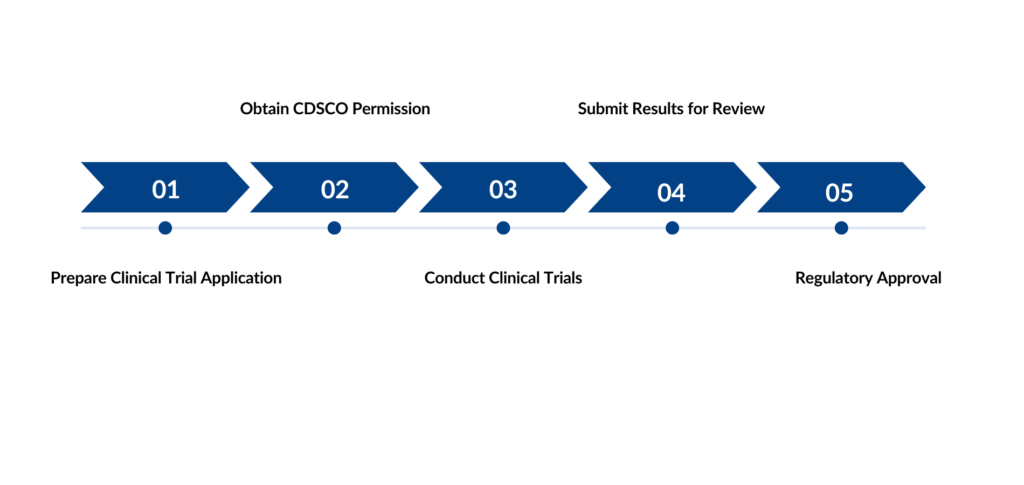
Clinical trials are a systematic study of new drug(s) in human subjects to generate data on clinical, pharmacological, and/or adverse effects with the goal of determining the safety and/or efficacy of the new drug. In India, clinical trials are governed by the New Drugs and Clinical Trials Rules, 2019, which require approval from the Central Drugs Standard Control Organization (CDSCO) and the respective ethics committees.
For new drugs, trials are essential before approval for manufacturing, import, or marketing in India. Depending on the origin of the drug, the clinical trial process may involve conducting trials in India starting from Phase I, or Phase I data generated elsewhere may be submitted for approval before conducting further trials in India.

Steps for Clinical Evaluation/Trial Approval
UDI (Unique Device Identification) Registration
The FDA’s Unique Device Identification (UDI) system helps identify and track medical devices in the U.S. market. Device manufacturers or brand owners are required to include a UDI on device labels and packages and submit device details to the FDA Global Unique Device Identification Database (GUDID).
At Accorpmed, we assist manufacturers in navigating the UDI registration process, ensuring that all medical devices are properly labelled, compliant with FDA regulations, and submitted to the GUDID. Our team helps streamline the entire process, from obtaining necessary identifiers to managing device information on the FDA database.

Steps for UDI (Unique Device Identification) Registration
- Obtain DUNS Number: Validate labeler information.
- Request GUDID Account: Assign a regulatory contact and request access.
- Assign UDIs: Verify GMDN standard codes and create UDI codes (Device Identifier and Production Identifier).
- Submit to GUDID: Upload device information, including UDI and production details.
Required Documents for UDI (Unique Device Identification) Registration
- Device Classification Report
- UDI Assignment Details
- Labeling Compliance Documents
Product Recall (Procedure Compliance)
A product recall ensures that defective or harmful medical devices are removed from the market to protect public health. Companies are required to promptly notify authorities, take corrective actions, and ensure compliance with recall procedures. At Accorpmed, we guide manufacturers through each stage of the recall process, ensuring effective execution, from initiation to post-recall monitoring.

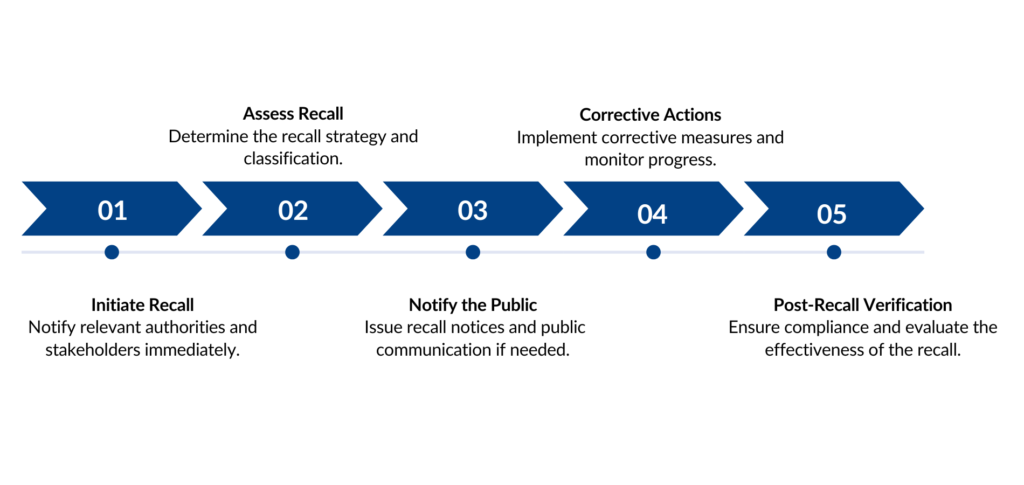
Stages of Recall Procedure
Receipt of Problem Report
Recall Initiation
Assessment of Recall
Recall Execution
Progress Reports
Evaluation
Required Information for Recall Assessment
Problem Details
Contact info of the reporter, date, nature of the issue, and test results.
Product Details
Name, batch number, manufacturing info, local & overseas distribution lists.
Health Hazard Evaluation
Type of hazard, proposed action, classification level, and alternative product availability.
Steps Product Recall Procedure Compliance

AccorpMed streamlines medical compliance with expert licensing and certification solutions. Trusted by 100+ healthcare businesses, we ensure hassle-free approvals for land, product, labor, and distribution licenses.
Contact Information
support@accorpmed.com
+91 99682 97717
909, ITL Twin Tower, B-9, Netaji Subhash Place, Pitampura, Delhi-110034 (INDIA)
2025-26 © accorpmed.com | All Rights Reserved